Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society.
Melzer, Nico, et al.
Journal of neurology 263 (2016): 1473-1494.
1. 背景
重症筋無力症 (MG) は、自己免疫性の抗体介在性神経筋シナプス伝達障害である。これは、(a) 神経筋接合部 (NMJ) における自己抗体蓄積が検出されていること、(b) MG患者の自己抗体を齧歯類に注入するとMG症状を引き起こすこと、(c) 動物に自己抗原を能動免疫すると疾患を引き起こすこと、(d) 抗体除去療法がMG症状の重症度を軽減すること、によって支持されている。
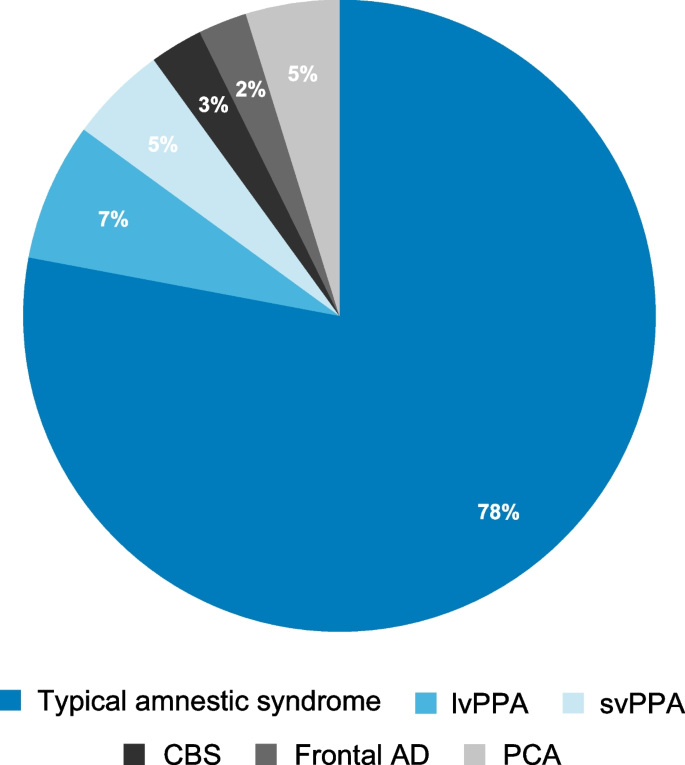
MGの発症率は1,000,000人に0.25から2.0人である。効果的な治療戦略と正常な寿命に起因して、MGの有病率はこの数年で72:1,000,000 (range: 15-179) にまで増加した。10%の患者は小児または思春期である。MGには家族性リスクがある。患者のきょうだいや1親等親族はMGの発症リスクが4.5%であり、この疾患の遺伝的素因を反映している。
MGの臨床的特徴は、眼筋群、球および (近位) 四肢骨格筋群に影響を及ぼす、変動性の疲労性および筋力低下である。実用的な臨床分類では、純粋な眼筋型筋無力症と、軽症、中等症および重症の全身型筋無力症が区別される。眼筋型筋無力症は、眼瞼挙筋を含む外眼筋にのみ発現し、眼瞼下垂および複視を呈する。眼瞼下垂と複視は1日のうちで一過性、変動性、または進行性である。10-20%の患者だけが、持続的に外眼筋に限局した易疲労性と筋力低下を呈する。患者の大部分は、発症後24ヵ月以内に全身性の易疲労性と筋力低下に移行する。全身型筋無力症は、その重症度とは無関係に、外眼筋以外の筋群の臨床症状として定義される。
易疲労性と筋力低下の変動性は、3Hzの周波数で副神経または顔面神経を反復的に最大上刺激した際に、最初の刺激と比較して5回目 (4回目の施設もある) のCMAPの振幅および/または曲線下面積が減衰していることによって示される。周波数30Hzの神経刺激や、等尺性筋収縮前後の単回刺激で、CMAPの振幅や曲線下面積に漸増的な反応がないことは、神経筋伝達障害のシナプス後性の性質を証明している。単線維筋電図では、不安定な神経筋伝達を反映して、一般的にジッターの増加や断続的な伝導ブロックがみられる。
2. 各MGサブタイプごとの疫学、免疫学、遺伝学的特徴
臨床、疫学、免疫学、遺伝学的な発見と胸腺病理に基づき、MGはさらに下位分類される (表1): 純粋眼筋型MG (OMG) は、若年発症 (<45歳, early-onset MG, EOMG) または後期発症 (>45歳, late-onset MG, LOMG) の全身型MGと区別される。EOMGはしばしば胸腺のリンパ濾胞性過形成と関連付けられ、LOMGは胸腺の年齢依存性の萎縮によって特徴づけられる。これとは対照的に、10-15%の患者は胸腺腫を有する (thymoma-associated MG, TAMG)。


MGは、自己抗体によって神経筋終板における機能的な骨格筋ニコチン性アセチルコリン受容体 (AChR) が減少したり、終板の構造変化が生じたりすることで発症する。約85%では、AChRそのものに対する自己抗体が検出される。AChRは五量体リガンド依存性一価カチオンチャネルであり、相同なα、β、γ、δ、εサブユニットの化学量論によって定義された2形態で存在する: 胎児型AChRはα2βδγサブユニット構成、成人型AChRはα2βδεサブユニット構成である。αサブユニットは2つの機能的に重要なドメインを有している: (a) リガンド結合に重要な細胞外システインループ、(b) MIR (main immunogenic region) と呼ばれる大部分のAChR自己抗体が結合する細胞外配列。
発生の過程で筋が神経支配を受け、胎児型のAChRのγサブユニットがεサブユニットに置換され成人型のAChRとなる。正常では、フォールディングされたサブユニットから成る機能的なAChRを発現しているのは骨格筋と胸腺筋様細胞のみである。正常胸腺では、成人型と胎児型のAChRは神経支配を受けていない胸腺筋様細胞において発現しており、この細胞は筋蛋白に対する中枢性免疫寛容の導入に重要であると考えられている。
さらに、フォールディングされていないAChRサブユニット (機能的チャネル全体ではない) は胸腺上皮細胞においても発現しており、これはAIRE (autoimmune regulator) の部分的制御下にある。AIREはMHC分子による分化中のT細胞に対するAChRペプチドの提示を制御しており、正常ではAChRに対する免疫寛容をサポートしている。
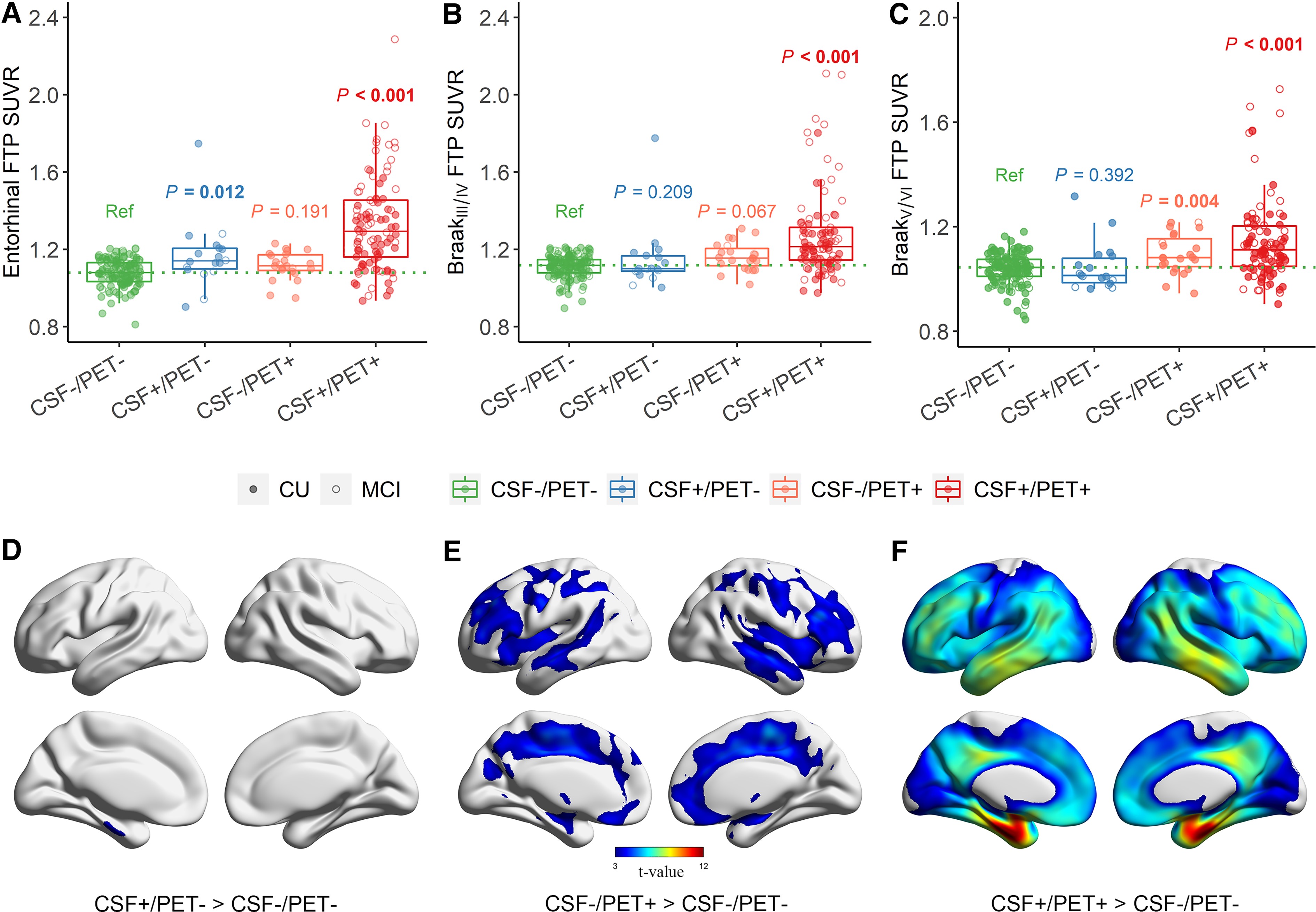
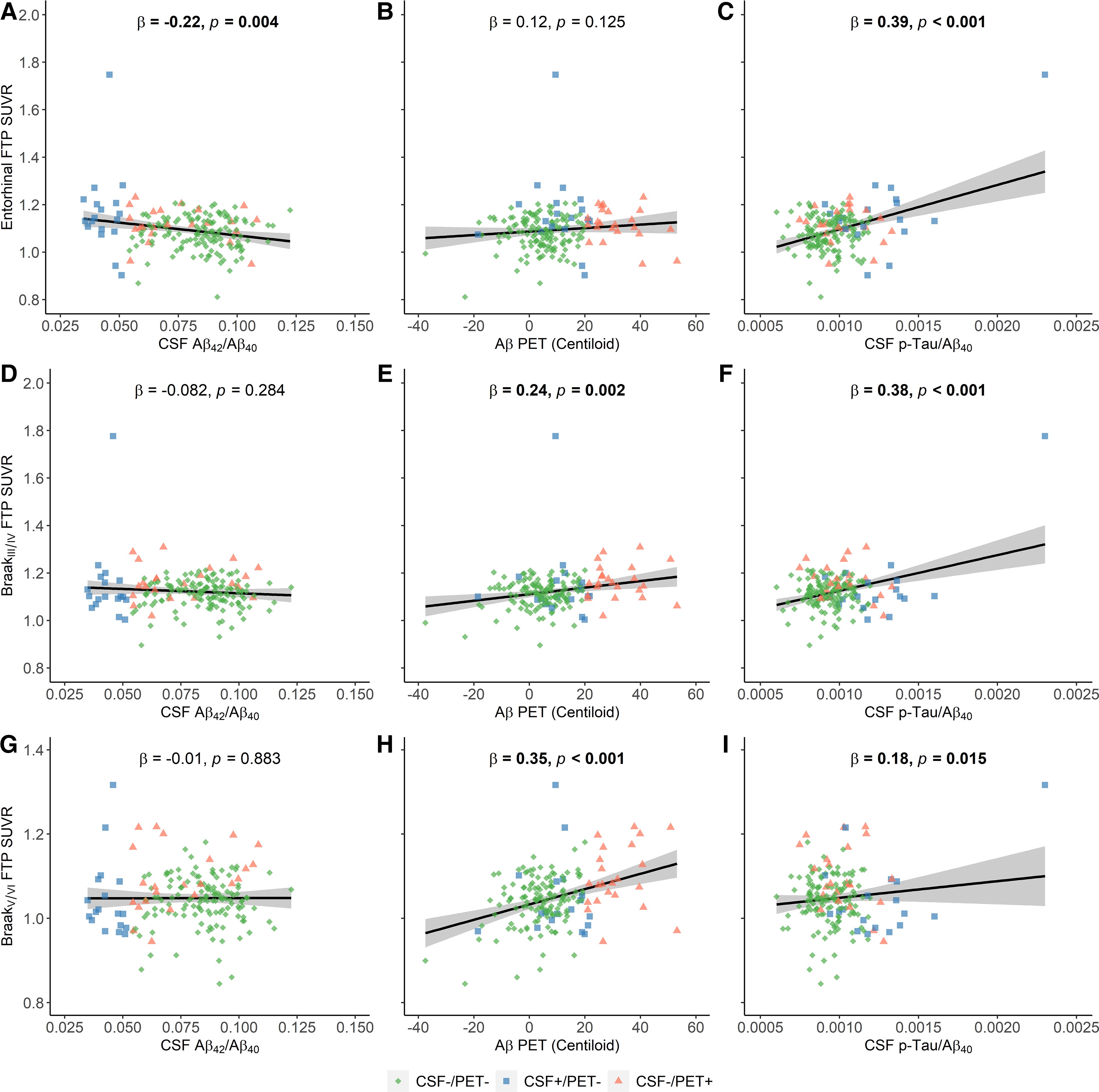
AChRに対する抗体は、部分的には低親和性抗体 (5%) であり、高親和性抗体 (80%) とは対照的に、cell-based assay (CBA) でのみ細胞表面上のクラスターとして検出されるが、標準的なラジオイムノアッセイ (RIA) では検出されない。AChRのMIRに対する抗体のレベルは、補体結合性のIgG1および3タイプであり、疾患の重症度と相関している。これらの抗体 (図1、2) は、(a) 受容体をブロックし、(b) 受容体の内在化を引き起こし、膜内で利用可能な受容体の数を減少させる可能性がある。さらに、(c) 補体カスケードの活性化により、終板構造が破壊されてシナプス間隙 (i.e. シナプス前アセチルコリン放出部位とシナプス後終板との間の距離) が広がり、アセチルコリン分子が放出部位から受容体へ拡散する距離が長くなる。
発症当初には、MGにおけるAChRに対する液性自己免疫反応は通常AChRのαサブユニットの単一エピトープに集中していることが多い。しかし、疾患経過とともに、筋や胸腺筋様細胞における自然AChRの二次関与 (i.e. プロフェッショナルなプロセシングおよび抗原提示) によって、この反応はαサブユニット内の他のエピトープや、さらには他のサブユニットまたは抗原へと広がる可能性がある。
MuSKに対する自己抗体は、補体非結合性のIgG4タイプであり、LRP4 (low-density lipoprotein receptoer-related protein 4) とMuSKの相互作用を阻害し、agrinによる神経筋接合部アーキテクチャを乱す。LRP4に対する自己抗体は主に補体結合性のIgG1および2タイプであり、LRP4-agrin相互作用を阻害して筋細胞におけるAChRクラスタリングを変化させる。Agrinに対する自己抗体はagrinによるMuSKのリン酸化とAChRクラスタリングを阻害する。Agrinに対する自己抗体のIgGサブタイプ分類は未だ研究されていない。
AChR抗体とMuSK抗体は基本的に同一患者で共存することはないが、同一患者でLRP4抗体とAChR抗体またはMuSK抗体が重複することは報告されている。さらに、agrin抗体もMuSK、LRP4、AChRに対する抗体と同時に検出されることが報告されており、agrin陽性MG症例では複数の神経筋接合部蛋白質に対する自己抗体が陽性になりやすいことを示している。
3. 個々のMGサブタイプにおける胸腺の役割
MGは、明確な免疫遺伝学的特徴を有する自己免疫疾患として現れるか、胸腺の腫瘍と関連する腫瘍随伴症候群として現れるが、他の悪性腫瘍と関連することは稀である。胸腺は、AChR抗体を有する患者の大部分 (OMG、EOMG、LOMGおよびTAMGの患者の大部分; 表1) において病理学的変化を示し、この変化は、中枢性および末梢性寛容の障害とMGの免疫病理学的発症の開始にとって中心的に重要であるように思われる (図1、2)。LRP4 抗体を有する一部の患者においても、胸腺の病理学的変化が報告されている。しかし、胸腺腫やその他の胸腺病変がMuSK抗体によるMGと関連することは稀であり、またagrin抗体によるMGにおける胸腺の変化に関するデータはまだ報告されていない。
自己抗原を発現または提示する胸腺間質細胞 (上皮細胞、樹状細胞、筋様細胞) と発育中の胸腺細胞との相互作用を介して、自己反応性T細胞のほぼ完全な排除が通常達成される。そして自己寛容性T細胞は分化を続け、最終的に末梢に排出される。生理的条件下では、胸腺の大部分は胸腺細胞 (i.e. 発育中のT細胞) と間質細胞を含み、B細胞の数は非常に少ない。
EOMG患者の約70% (図1) は、リンパ濾胞過形成 (lymphofollicular hyperplasia, LFH)、すなわち胸腺内にリンパ濾胞と胚中心を伴う胸腺炎を示す。未知の最初の "引き金 "に続いて、MHC-class IIを発現する過形成性の胸腺上皮細胞 (thymic epithelial cells, TECs) は、フォールディングされていないAChRサブユニットを提示し、自己反応性CD4+ T細胞を活性化するようである。プライミングされたT細胞によって惹起された初期抗体は、フォールディングされたAChRを発現している近傍の筋様細胞を攻撃し、補体を活性化してAChR/免疫複合体を放出すると考えられている。これらのAChR/免疫複合体は、今度は自己反応性CD4+ T細胞のさらなる活性化を促す専門の抗原提示細胞を活性化し、B細胞受容体の親和性成熟を伴う自己反応性B細胞のさらなる活性化と拡大を引き起こし、高親和性の後期AChR抗体の産生とそれに続くエピトープの多様化につながる。
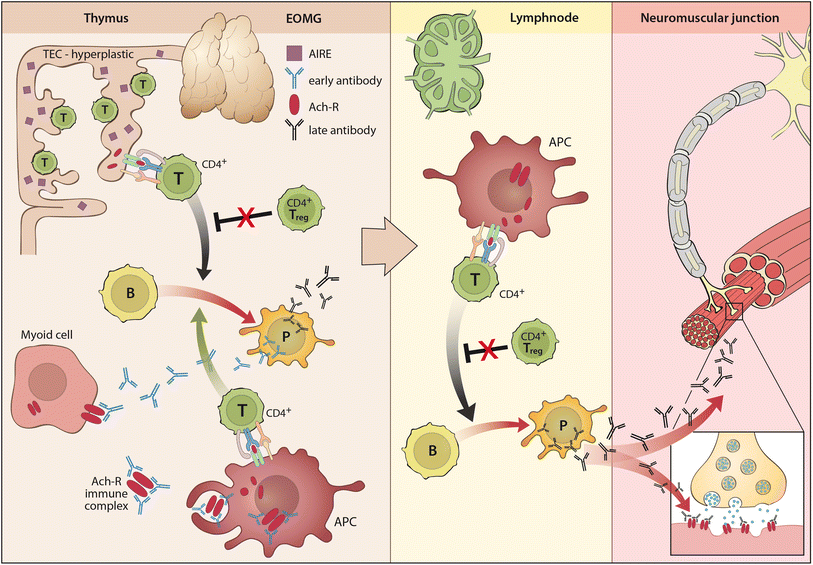
図1. リンパ濾胞過形成 (LFH) を伴う若年発症MG (EOMG) の発症機序
EOMGの胸腺と血液中に存在する制御性T細胞の機能不全が、EOMGにおける自己免疫炎症プロセスを自己増殖させているようである。おそらく、胸腺で開始された自己免疫過程は、後に末梢リンパ組織へ波及し、局所リンパ節に存在する骨格筋由来のAChR/免疫複合体や、機能不全に陥った制御性T細胞が、EOMGの維持に寄与しているのであろう。
次に、MG患者の10-15%が胸腺腫を有し、胸腺腫患者の約30%がTAMGを有する (図2)。胸腺腫は胸腺上皮細胞 (TEC) の新生物であり、通常、皮質と髄質が混在している。リンパ球の含有量および上皮細胞の特徴により、現在の組織学的分類ではA型、AB型、B1型、B2型、およびB3型の胸腺腫を区別している。すべての胸腺腫の95%以上 (まれなA型およびB3型を除く) は、骨髄前駆細胞からポリクローナルCD4+およびCD8+胸腺細胞を生成する。このような胸腺造血は、TAMGの病因において中心的な役割を果たしている: MG陽性胸腺腫は、成熟CD4+ CD45RA+細胞を大量に生成し、血液中に排出するが、MG陰性胸腺腫は生成しない。したがって、造血不全の胸腺がんはMGとは関連しない。
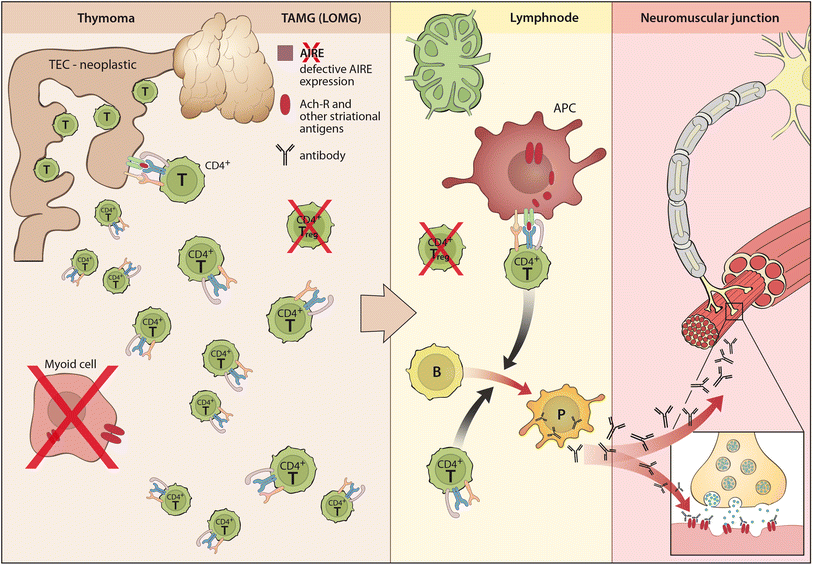
図2. 胸腺腫関連 (および後期発症) MG (TAMG, LOMG) の発症機序
しかし、胸腺腫における胸腺造血は、自己免疫を好む状況で発生する: 胸腺腫では、胸腺上皮細胞において末梢組織自己抗原 (AChR αサブユニットを含む) を手当たり次第に発現させる役目を持つAIREの発現が低下しており、さらに胸腺筋様細胞が減少または消失している。さらに、腫瘍性の上皮細胞では、MHC-class IIの発現レベルは低下している一方で、titinを含む横紋筋抗原のエピトープや、様々なAChRサブユニット (受容体全体ではない) を発現している。これらの腫瘍性上皮細胞の特性変化は、成熟胸腺細胞の (正および負の) 選択と成熟T細胞の活性状態と深く関連している可能性がある。さらに、AIREの発現低下と同調して、こうした変化は制御性T細胞の産生障害をきたす。同時に、これらの胸腺微小環境の変化は、ナイーブおよびプレプライミングされた自己反応性T細胞の多くを末梢に送り出すことを好み、これらが徐々に末梢に存在するより寛容なナイーブT細胞レパトアを置換するものと思われる。これらのT細胞は末梢リンパ組織において病原性B細胞応答を刺激するはずである。これは基本的に胸腺切除前に起こるものであるが、胸腺切除後でも稀に起こる現象である。胸腺腫が末梢免疫システムに与えるこの影響は、TAMGが一度発生すると胸腺腫を完全切除しても維持される理由を説明している。そして、領域リンパ節において制御性T細胞の非存在下で処理された骨格筋由来AChR/自己抗体の複合体は、TAMGを永続化させると考えられる。
EOMGの自己免疫は主にAChRに対するものであるが、TAMG患者の自己抗体標的のスペクトラムはより広い可能性がある (表1)。自己抗原のスペクトラムには、以下が含まれると思われる: (a) 骨格筋AChRや電位依存性Ca2+およびK+チャネル、その他のリガンド依存性神経伝達物質受容体を含むリガンドおよび電位依存性イオンチャネル、そしてこれらと複合体を形成する蛋白質、(b) titinおよびリアノジン受容体 (RyR) などの横紋筋抗原、(c) IFN-α、IFN-ω、IL-12を含むサイトカイン。TAMGにおけるこの自己抗原の拡張スペクトラムは、MG患者に起きやすいその他の自己免疫疾患の発症をも説明可能である。
LOMG (図2) の患者では、胸腺は退縮し萎縮している。正常加齢胸腺のリンパ上皮組織は徐々に脂肪置換されるが、成人になっても残存間質から少数ながらT細胞が送り出されている可能性がある。しかし、LOMGで残存リンパ上皮組織が拡張や浸潤といった徴候を示すことはほとんどなく、形態学的解析によれば、LOMG患者の胸腺と正常胸腺には有意な違いは認められなかった。胸腺筋様細胞は、LOMGではまばらな傾向があり、加齢とともに減少し、60~70歳の間にほぼ欠乏状態に達するが、かなりの個人差がある。加えて、AIRE陽性細胞の数も同様に減少するようであるが、LOMG胸腺と年齢をマッチさせた対照との間に明確な差はない。
LOMG患者の免疫学的特徴は、TAMGとかなり類似している (表1): (a) titinに対する自己抗体は70%の患者 (特に60歳以上) で認められ、それ以外の患者でもRyR抗体が認められることがある。(b) およそ40%がIFNαかつ/またはIL-12に対する中和抗体を有する。(c) 50%以上がTAMG患者と末梢性T細胞レパトアを共有している。したがって、LOMGの加齢胸腺における免疫学的異常は、明らかな新生物を伴わなくとも胸腺腫のそれと類似しているように見え、そして非寛容T細胞を末梢に送り出し、さらに活性化すらさせている可能性がある。診断時点のLOMG患者では、末梢へのナイーブT細胞の送り出しが増えているわけではない。しかし、MGの診断がつく前の段階で小さな胸腺腫が存在し、それが自然に縮小してしまった可能性はある。さらに、ほぼAIRE陰性の萎縮胸腺において、かつ筋様細胞がほとんど存在しない中で生成されたAChRおよびtitinに反応性のT細胞の小さな集団は、末梢に送り出されてから活性化されてLOMGを惹起している可能性があり、また病原性T細胞集団はLOMGが発症するずっと前から末梢に蓄積していた可能性もある (i.e. 胸腺切除術後にTAMGを発症する稀な患者における機序と同様のものを想定している)。一度免疫機序が開始されてしまえば、LOMGはEOMGおよびTAMGに関して上で述べたように、筋灌流リンパ節におけるAChR/自己抗体複合体による刺激を受けながら自己永続的に持続する。
4. MGの治療戦略
自己抗体の状態とは無関係に、MGの治療は同じ原則に従って行われる。治療戦略は、(a) 神経筋伝達を促進する対症療法と、(b) MGの根底にある病的な免疫反応を標的とする免疫抑制療法に区別することができる。
4-1. 対症療法
ピリドスチグミン臭化物などのアセチルコリンエステラーゼ阻害剤 (AChEI) は、最も一般的な対症療法である。これらの薬剤の臨床的有効性は、電気生理学的測定により証明されている。しかし、MGの治療におけるこれらの薬剤の広範な適用は、対照のない観察研究、症例シリーズ、および良好な臨床実践に基づいている。倫理的な理由から、これらの化合物の臨床的有効性に関するプラセボ対照試験は禁止されている。
MuSK抗体関連MGの患者は、通常、AChEIによる治療に対して、nAChR抗体関連MGの患者よりも反応が悪い。このような症例では、症状コントロールのために高用量が必要となり、しばしば全身性の副作用が増加する。
ピリドスチグミン臭化物は現在、MGの経口長期治療に使用されている。通常、300mg/日以下の投与量ではコリン作動性の副作用は生じない。しかし、静脈内投与では、気管支痙攣および分泌過多、筋力低下の増悪、腹部痙攣、尿意切迫、唾液分泌過多および発汗、徐脈および房室ブロック、縮瞳などのコリン作動性副作用が発現することがあり、コリン作動性中毒となる。ピリドスチグミン臭化物の静脈内投与(最大24mg/日)には、常に継続的なモニタリングが必要である。すべての投与方法において最も頻度の高い全身性の副作用は、胃腸障害、下痢および腹部痙攣 (約30%)、唾液分泌過多 (約6%)、発汗 (約4%)、徐脈および房室ブロック (約1%) である。
胃腸の副作用を伴う臭化物不耐症の場合は、ピリドスチグミン臭化物の経口投与の代わりに、アンベノニウム塩化物の経口投与が使用できる。
4-2. 免疫療法
全身型MGに対する免疫抑制剤の有効性は一般に認められている。さらに、純粋な眼筋無力症の患者は、免疫抑制下にある場合、全身型MGに進行する割合が減少することが示されている。
しかし、より大規模なランダム化比較試験で検証され、MG患者に使用できる明確なクラスIのエビデンスが得られている免疫抑制薬はわずかである。ランダム化比較試験の中には、免疫抑制剤の臨床的効果が証明されないものさえある。しかし、これらの試験には、後述する方法論的な弱点がある。
これとは対照的に、免疫療法を中止する期間と基準に関する臨床試験からの証拠は乏しい。一般的に、数年間の安定した臨床的寛解の後、免疫抑制剤治療を長期に漸減することは可能であると思われる。特に臨床的に不安定な状況で免疫抑制を突然中止すると、筋無力症状の急激な悪化や筋無力症クリーゼを引き起こす可能性がある。実際、ほとんどの患者は終生免疫抑制療法を必要とし、日和見感染症、リンパ腫、その他の重篤な治療関連副作用を引き起こしやすい。対症療法と免疫療法の全体的な目的は、完全またはほぼ完全な臨床的寛解である。
4-2-1. 基本的免疫治療
MGの免疫抑制における第一選択薬は、副腎皮質ステロイドとアザチオプリンである。その他の免疫抑制剤も、禁忌や不耐性、臨床的疾患制御不十分の際には使用可能である。第二選択の免疫抑制剤は、シクロスポリンA (ポジティブな対照試験が1つ存在する)、メトトレキサート (ポジティブな対照試験が1つ存在する)、ミコフェノール酸モフェチル (フォローアップ期間がかなり短いネガティブな対照試験が2つ存在する)、タクロリムス (フォローアップ期間がかなり短いネガティブな対照試験が1つ存在する)。
4-2-2. 副腎皮質ステロイド
後ろ向き研究では、プレドニゾン、プレドニゾロン、メチルプレドニゾロンなどの副腎皮質ステロイド (glucocorticosteroid, GCS) は、70-80%の患者で複数週から月の期間内に臨床症状を改善することが示されている。副作用の観点から、長期間の経口GCSはアザチオプリン、シクロスポリンA、メトトレキサート、ミコフェノール酸モフェチル、タクロリムスなどのステロイドスペアリング免疫抑制剤と組み合わされることが多い。GCS治療の開始から数日以内は、筋無力症状の一過性の増悪をきたすことがあり、これは特に球症状に強い症状を持つ患者で起こりやすい。
臨床現場では、3つの異なる用量レジメンが使われている。
1. 初回投与量はプレドニン換算で10-20mg/日、安定した寛解に達するまで1週間ごとに5mg/日ずつ増量する (約1mg/日/kg体重)。利点: 治療初期における筋無力症状の一過性の悪化を予防できる。欠点: 臨床的改善が遅い。
2. 安定した臨床的寛解が得られるまで、ステロイドスペアリング免疫抑制剤と併用しながらプレドニゾン換算で1-1.5mg/日/kg体重から開始し、その後、GCS療法の完全中止を目標に、4週間ごとに5mg/日の減量を行う。利点: 迅速な臨床的改善。欠点: 約10%の患者で、治療開始後数日間に筋無力症状が一過性に悪化する。
3. 500-2000mg/日を3-5日間連続投与するメチルプレドニゾロン静注パルス療法を行い、その後経口的に漸減する。この治療法は、GCSの直接的な膜効果により一過性に筋無力症状を悪化させ、ひいては筋無力クリーゼを誘発する可能性がある。さらに、急性のステロイドミオパチーが、このような状況における臨床全体の悪化に寄与する場合もある。したがって、この治療レジメンは、明らかな筋無力症クリーゼの場合にのみ、また、中間または集中治療環境において、血漿交換療法、免疫吸着または免疫グロブリン静注療法と組み合わせて使用される。
GCSの副作用の数と重症度は、投与期間と累積投与量とともに増加する。特に、糖尿病など他の合併症を有する患者は、特別なリスクがある。プレドニゾロン換算で7.5mgを超える投与量で治療期間が3ヵ月を超えると予想される場合は、骨粗鬆症を予防するためにカルシウム (1000~1500mg/日) とビタミンD (400~800IE/日) を投与すべきである。ビタミンD濃度は、このような治療を開始する前に測定し、全体を通してコントロールすべきである。閉経後の女性では、ビスフォスフォネートを用いてGCS誘発性骨粗鬆症を予防することができる。男性におけるGCS誘発性骨粗鬆症と骨折に対するビスフォスフォネートの予防効果に関するエビデンスは、現在のところ一般的な推奨には十分ではない。さらに、プロトンポンプ阻害薬や他の薬剤を用いた胃の保護が正当化されるかもしれない。
副作用を軽減するために、低用量域での長期投与中にGCSを毎日投与から交互投与に切り替える施設もあるが、系統的なデータがない以上、個々の患者においてその有用性を検証する必要がある。
4-2-3. アザチオプリン
アザチオプリン (AZA) は、MGの治療に最も頻繁に使用される免疫抑制剤である。アザチオプリンはプリンアナログであり、速やかに代謝され、細胞毒性および免疫抑制誘導体である6-メルカプトプリンとチオイノシン酸に変化する。後者はプリン合成を阻害するため、活性化と増殖を障害し、ヌクレオチドのサルベージのための代謝経路がないT細胞とB細胞のアポトーシスを引き起こす。治療は2-3mg/日/kg体重で開始され、臨床的に安定した寛解が得られた場合には、治療期間中に約2.5mg/日/kg体重、さらに1mg/日/kg体重まで減量される。治療効果は数ヵ月以内には期待できない。しかし、長期治療中のAZAのステロイドスペアリング効果は証明されている。AZAとプレドニゾロンの併用は、それぞれの単剤療法に比べて寛解状態が長く、副作用が少ないため、より効果的である。10-20%の患者では、AZAとGCSの併用で十分な臨床的安定化または寛解が得られず、プレドニゾロン換算で7.5mg/日を超えるGCSの投与が必要となり、長期的には他の免疫抑制治療戦略が必要となる (治療抵抗性)。
AZAの突然の中止は、完全かつ安定した臨床的寛解状態にある患者においても、筋無力クリーゼに至るまでの筋無力症状の再発を誘発することがある。
AZAで治療された患者の約80%において、赤血球の平均赤血球容積 (MCV) の増加が観察され、これは治療反応者で非反応者に比べてより顕著であり、頻度も高い。AZAはその作用機序により、定常状態では可逆的なリンパ球減少を引き起こす可能性がある。絶対リンパ球数は600-1200/μlの範囲であるべきで、総白血球数は3500/μlをはるかに超えていなければならない。
AZAはプロドラッグであり、グルタチオン-s-トランスフェラーゼによって活性代謝物の6-メルカプトプリンと1-メチル-4-ニトロ-5-チオイミダゾールに代謝される。6-メルカプトプリンはキサンチンオキシダーゼまたはチオプリン-S-メチルトランスフェラーゼ (TPMT) によってさらに代謝される。アロプリノールなどのキサンチンオキシダーゼ阻害剤はアザチオプリンの代謝を阻害する。この薬剤と同時に使用する場合には、骨髄毒性の副作用を防ぐため、アザチオプリンは標準用量の25% (すなわち、0.5-0.75mg/日/kg体重) に減量してのみ使用することができる。AZAが必要な場合には、キサンチンオキシダーゼ阻害剤の代わりに、ベンズブロマロンやプロベネシドなどの他の薬剤を尿酸値の低下に使用することができる。
少数の患者 (1%未満) では、吐き気や嘔吐、下痢を伴う胃腸障害、循環抑制のような急性の重篤な副作用が特異的な即時反応として起こり、患者がAZAによる治療を受けられなくなることがある。このような特異的反応を除外するために、AZAによる長期治療を開始する前に、このような副作用を検出するために50mgの単回経口投与試験を行うことが推奨される。
遺伝的にTPMT活性が低い場合、AZAは治療開始直後に予想外に強い骨髄抑制を引き起こす。治療開始前にTMTP活性またはTPMT遺伝子型の検査を行うことができる: (a) TMPT活性を完全に欠く患者 (頻度1:300) またはTPMT一塩基多型のホモ接合体はAZAで治療できない。しかし、この遺伝子型は非常にまれである (約0.5%)。この遺伝子型が上記の特異的即時型反応と関連しているかどうかは、現在のところ不明である。(b) TPMT活性が非常に高い場合、AZAは臨床効果をもたらすことなく急速に代謝される可能性がある。
10年未満のAZA治療による悪性腫瘍のリスク増加は明らかでない。MG患者では、まれに血液悪性腫瘍や日和見感染症の症例が報告されている。さらに、アザチオプリン投与下での皮膚角化症および皮膚癌の発生率の増加が報告されており、これはおそらく光線過敏症の増加によるものであろう。アザチオプリンによる長期治療を受けている患者には、定期的な皮膚検査が推奨される。汎発性疣贅や多発性基底腫が発生した場合には、AZA治療を減量するか、別の薬剤に変更しなければならない。強い太陽光線照射下で急性光毒性反応が数例報告されている。
4-2-4. シクロスポリンA
シクロスポリンA (CSA) は、プラセボ対照臨床試験において、MG 患者に対する有効性が証明され、クラスIのエビデンスが得られている。
シクロスポリンは、リンパ球の細胞質タンパク質であるシクロフィリンに結合し、抗原特異的リンパ球の活性化、分化、エフェクター機能の発揮に必要な、抗原受容体刺激後のカルシウム-カルモジュリンによるカルシニューリンの活性化を阻害する。
上記の臨床試験 (6mg/日/kg体重を2回に分けて投与するCSA単剤療法) と比較して、CSAは現在、GCSとの併用で、最初は3-4mg/日/kg体重、その後2-2.5mg/日/kg体重に減量して2回に分けて投与する低用量で臨床使用されている。治療モニタリングは血中濃度 (12時間の投与間隔終了時のトラフ値) で行う。AZAと比較すると、CSAの臨床効果はより速やかに、すなわちほとんどが4-6週間以内に発現する。しかし、CSAは投与量に応じて生じる副作用の範囲がはるかに広く、日和見感染、骨髄抑制、歯肉過形成、胃腸障害、高カリウム血症を伴う腎毒性 (治療中は腎機能のモニタリングが必要)、動脈性高血圧などがある。特殊な副作用としては、振戦、頭痛、てんかん発作の増加傾向、まれに可逆性後部白質脳症症候群などがある。
さらに、CSAは他の薬物との相互作用が多く、特に高齢で多疾患を合併している患者においては、厳重な薬物モニタリングが必要である。
4-2-5. メトトレキサート
メトトレキサート (MTX) は、MGの治療薬として数十年前から使用されている。MTXはジヒドロ葉酸還元酵素 (DHFR) をジヒドロ葉酸の約1000倍の親和性で競合的に阻害する。ジヒドロ葉酸還元酵素は、ジヒドロ葉酸から活性代謝物であるテトラヒドロ葉酸への変換を触媒し、プリンおよびピリミジンヌクレオチドのデノボ合成に関与する。したがって、メトトレキサートはDNA、RNA、タンパク質の合成を阻害し、リンパ球などの増殖を抑える。MTXの最も顕著な副作用は、肝毒性、潰瘍性口内炎、白血球減少、貧血、感染症、悪心・嘔吐、腹痛、急性肺炎、まれに肺線維症や腎不全などである。
MG患者に長期的に使用されているにもかかわらず、最近まで対照臨床試験が欠如していた。最近の臨床試験では、全身型MG患者24人を対象に、MTX (17.5mg/週) とAZA (2.5mg/日/kg体重) をステロイドスペアリング効果に関して比較し、治療期間2年以内で同等の効果を示した。MTXは、葉酸との併用で7.5-25mg/週の用量であるため、MGの治療における第二選択薬とみなすことができる。
他の薬剤との相互作用の頻度が低いため、特に高齢の多疾患合併患者では、CSAよりもMTXの方が望ましいと考えられる。
4-2-6. ミコフェノール酸モフェチル
ミコフェノール酸モフェチル (MMF) は、イノシン一リン酸脱水素酵素 (IMPDH) を非競合的に阻害するため、特にリンパ球の細胞増殖に必要なプリンヌクレオチドのデノボ合成を阻害する。
MMFの最も顕著な副作用は、慢性下痢、溶血性貧血、浮腫である。MMFによる治療で進行性多巣性白質脳症が数例報告されている。さらに、MMFには催奇形性があることが証明されている。したがって、妊娠を計画している場合には、少なくとも妊娠の4ヵ月前にはMMFの投与を中止しなければならない。予定外の妊娠の場合は、MMFの投与を直ちに中止し、その後、婦人科医のカウンセリングを受ける必要がある。
いくつかの観察コホート研究において、MMFによる治療でステロイドスペアリング効果を伴う臨床的改善が、1500-2000mg/日の投与量で、薬剤モニタリングのもとで認められている。しかし、2つの臨床第III相試験において、MMFは初期治療としてプレドニゾンより優れておらず、9ヶ月間調査してもステロイドをスペアする効果は認められなかった。MMFで起こることが知られている臨床効果の潜伏期間を考慮すると、2つの第III相試験における36週間以内という追跡期間はかなり短いように思われる。さらに、プレドニゾンの治療効果は予想外に良好であった。したがって、MMFの効果は、これらの試験の方法論上の問題から過小評価されているように思われる。一貫して、その後の非対照コホート研究でも、MMFの単独療法またはプレドニゾンとの併用療法の有益な効果は、6ヶ月の治療後に示された。
4-2-7. タクロリムス
タクロリムス (TCM) は、CSAと同様にカルシニューリン阻害剤であり、抗原特異的リンパ球の活性化、分化、リンパ球のエフェクター機能の発揮を効果的に阻害する。TCMの有効性はCSAの10-100倍である。副作用はCSAと同様で、強い用量依存性を示す。TCMは日本で開発され、MGの治療薬として認可されている。治療抵抗性のMG患者におけるTCM (3-5mg/日) の有益な臨床効果が、いくつかの公開臨床試験や小規模なケースシリーズで報告されている。79名のMG患者を対象とした多施設共同オープン・コホート研究において、低用量のTCM (0.1mg/日/kg体重) は、CSAとプレドニゾロンからなる併用療法に取って代わることができ、nAChR-Ab価の低下を含む良好な臨床的安定化をもたらした。経口プレドニゾロン療法 (10-20mg/日) を受けている臨床症状の軽微なMG患者80人を対象とした無作為プラセボ対照臨床試験では、28週間にわたってTCM (3mg/日) のステロイドスペアリング効果が検討された。経口プレドニゾロン療法は、TCM治療4週間後から段階的に減量された。しかし、追跡期間の最後の12週間の平均経口プレドニゾロン投与量に関しては、TCMとプラセボとの間に有意差は検出されなかった。研究集団と追跡期間が短かったため、この研究は、MG患者における中医学の長期的な有効性とGCS療法における不十分な治療効果に関する証拠をほとんど示していない。CSAと同様に、TCMは腎毒性および神経毒性を有し、複数の薬物相互作用を示す。
4-3. エスカレーション療法
4-3-1. リツキシマブとその他のモノクローナル抗体
多くの症例報告や症例シリーズが、モノクローナル抗CD20抗体であり循環Bリンパ球除去薬であるリツキシマブの重症治療抵抗性MGに対する臨床的有用性について報告している。しかし、ランダム化比較試験からのデータは未だ存在しない。
合計168人の患者を含む15個の非対照観察研究のメタ解析が近年行われた。125人の女性および43人の男性、91人の抗AChR抗体陽性および70人の抗MuSK抗体陽性患者 (7人はdouble seronegative) が対象とされた。フォローアップ期間中央値は、AChR抗体陽性患者については16カ月、MuSK抗体陽性患者については26カ月、double seronegative患者については12カ月であった。リツキシマブの用量レジメンは研究ごとに異なっていた: 137人が4 × 375 mg/m2、12人が500 mg on day 1 and 8、8人が1000 mg on day 1 and 15であった。残る11人の患者は異なる治療レジメンであった。全体の奏効率は83.9%であった。特に、MuSK抗体陽性患者では奏効率が高く (88.8%)、AChR抗体陽性患者 (80.4%) や double seronegative患者 (85.6%) よりも高かった。しかし、これらの違いは群間有意差には至らなかった。リツキシマブはすべての異なる治療レジメンで有効性が証明された。おそらくは、MuSKに対するIgG4抗体はほとんどがCD20陽性の短命な形質細胞によって産生されており、これはCD20陰性の長寿形質細胞から合成されると考えられているIgG1および3抗体を主体とする抗AChR抗体の性質と異なっている。この仮説は、リツキシマブがAChR抗体陽性例よりもMuSK抗体陽性例で高い有効性を示したことを説明する。リツキシマブに対して奏功したMuSK抗体陽性患者は、典型的には抗体価も低下したが、AChR抗体陽性患者では抗体価と臨床奏功性に解離がみられることがあった。
さらに、統計学的有意差の証明には至らなかったものの、罹病期間と奏効率の間には逆相関の傾向が観察された。この傾向も、罹病期間とともに増加する長寿形質細胞プールに帰属可能である。副作用は7/168人の患者で報告された (4.2%; 感染症が4人、B細胞除去の遷延が2人、心不全が1人)。治療関連の進行性多巣性白質脳症は報告されなかった。したがって、この非対照観察研究のメタ解析データは、難治性MGにおけるリツキシマブの使用をサポートする。しかし、多施設ランダム化比較試験は、MGにおけるリツキシマブの有効性の安全性を明確に確立するためには必要である。
エクリズマブはヒト化モノクローナル抗体であり、C5の酵素切断を特異的に阻害することによって終末補体複合体の形成をブロックする。近年のランダム化二重盲検プラセボ対照クロスオーバー第2相試験では、重症難治性全身型MG14例におけるエクリズマブの臨床的有効性が研究された。患者はエクリズマブ600mgまたはプラセボの毎週投与を4回受け (導入フェイズ)、さらに追加でエクリズマブ900mgまたはプラセボの隔週投与を受けた (維持フェイズ)。エクリズマブで16週間治療された患者7人中の6人 (86%) が主要エンドポイントであるQMGスコアの3点低下を達成した。さらに、平均QMG総スコアの総変化はエクリズマブとプラセボで有意に異なり、平均QMG総スコアのベースラインからの総変化もエクリズマブとプラセボで有意に異なっていた。エクリズマブに対する忍容性は高かった。したがって、この試験は、重症難治性全身型MGに対するエクリズマブの役割をサポートしている。第3相試験が現在行われている。
多発性硬化症で幅広く用いられているその他のモノクローナル抗体治療 (抗CD52/alemtuzumab、抗IL2R/daclizumab) の臨床経験はMGでは未だ少数であり、個々の稀な例に関してのみ考慮されるべきである。
4-3-2. シクロフォスファミド
シクロホスファミド (CPP) は、DNAにアルキル基を付加するナイトロジェンマスタードのアルキル化剤である。これは、鎖内および鎖間のDNA架橋を形成することにより、DNAの複製を阻害する。CPPは、標準治療に失敗した重症のMGの症例に適用することができる。さらに、CPPは、いくつかの小規模な対照症例シリーズによれば、反復免疫吸着療法や血漿交換療法を必要とする患者にも適用可能である。生命を脅かすような重篤な治療抵抗性のMGでは、いくつかの治療レジメンに肯定的な臨床エビデンスが存在するため、CPPを最後の手段として使用することができる:
1. CPPパルス療法:前向き無作為化二重盲検試験に基づき、メスナとの併用療法で寛解まで4週間ごとに500mg/m2体表面積を投与する。
2. 免疫・骨髄除去CPP療法: 50mg/day/kg体重を4日間連続で投与した後、小規模の症例研究に基づきGCSFを投与するか、自家または同種幹細胞移植を行う。
累積投与量と治療期間は、両性における妊孕性障害のリスクと悪性腫瘍のリスク (累積投与量と治療期間によって異なるが、約1%) の増加のため、記録されるべきである。単回投与の場合、累積投与量は50-70gに達する。これらの値は、CPPの静脈内投与に比べ、経口投与の方がはるかに早く到達するため、前者の投与経路が望ましい。CPP療法の合併症としては、悪性腫瘍、肺線維症、心筋症、皮膚線維腫などがある。
MGの治療法の概要については、表2を参照されたい。


4-4. インターベンション療法
以下の治療方法は、筋無力症クリーゼの予防と治療、および妊娠中の不安定なMGや重度の機能障害をきたす治療抵抗性筋無力症に対する治療として適用されるものである。
4-4-1. 経静脈免疫グロブリン (IVIG)
経静脈免疫グロブリン (IVIG) は数千の健常ドナーから得られたポリクローナル免疫グロブリンから成る。IVIGが自己免疫炎症を抑制する詳細なメカニズムは明確には確立していないが、FabまたはFcフラグメントを介した数多くの分子効果を含んでいると考えられる。副作用には頭痛、高血圧、アレルギー/アナフィラキシー反応 (特にIgA欠損患者)、皮膚炎、感染 (HIVまたはウイルス性肝炎)、IVIGの高い膠質浸透圧に起因する体液負荷過剰による肺水腫、静脈血栓症、無菌性髄膜炎、溶血がある。
IVIGは、0.4g/日/kg体重を5日間連続で投与する、あるいは1g/日/kg体重を2日間連続で投与する。IVIGは、筋無力症クリーゼ時の人工呼吸時間の短縮において、血漿交換や免疫吸着と同様の効果があることが示されている。
さらに、IVIGは、手術 (胸腺切除術を含む) の前や、重症のMGの場合、高用量GCSパルス静注療法を開始する前の臨床的安定化のために使用することができる。いくつかの非盲検臨床試験における臨床的奏効率は80%であった。
筋無力症クリーゼ以外の状況における導入療法または維持療法としてのIVIGの使用 (単独または免疫抑制剤の上乗せ療法) については、ランダム化比較試験のデータはない。しかし、IVIGはこの目的のために、長期間にわたって使用されることがある (最初は5×0.4g/日/kg体重、その後は4-8週間ごとに1×0.4g/日/kg体重)。治療抵抗性の筋無力症状を有する患者は、IVIGによる長期治療が有効であるようである。IVIGは、他の免疫抑制剤 (特にGCS) に禁忌のあるMG患者にも使用できる。
4-4-2. 血漿交換と免疫吸着
治療的血漿交換 (PE) により、血漿は血液成分から分離され、代用液と交換される。PEは血漿全体を除去する非特異的治療法である。治療効果は、自己抗体を含む循環病原性免疫因子の除去に基づいている。これとは対照的に、免疫吸着 (IA) は、特異的なマトリックス (e.g. プロテインAやトリプトファン) に結合してIgG抗体を除去する、より選択的な手法である。
血漿交換は、筋無力症クリーゼの治療法として成功している。さらに、治療抵抗性の場合、手術前 (胸腺切除術を含む) の患者の安定化、または重症MGの場合、高用量のGCSパルス療法を開始する前に、プラズマフェレシスを使用することができる。通常、臨床的寛解に達するまで、6-8回の治療 (1日おきに血漿量の1-1.5倍量の治療) を行う。免疫抑制を伴わなけれ ば、病原性自己抗体の (再) 合成により、臨床効果は数週間しか持続しない。一回の治療ごとに、ヒトアルブミンの補充が必要であり、二次性免疫グロブリン欠乏症 (IgG<150mg/dl) の場合は、多価IgGによる補充が可能である。凝固因子の枯渇も治療頻度を制限するため、他の抗凝固薬による治療を行う場合には考慮する必要がある。筋無力症クリーゼでは、血漿交換とIVIGは同等の効果があり、同等に使用できるようである。ランダム化比較試験では、両治療戦略の間に有意差は認められなかった。さらに、対照クロスオーバー研究と後ろ向きコホート研究でも、これらの治療法の間に有意差は認められなかった。中等度から重度のMG (QMGS > 10.5) で臨床的増悪を認めた84人の患者を対象とした最近の比較試験でも、60日間の追跡調査期間中、主要評価項目であるQMGSの低下 (IVIGでは69%、血漿交換では65%) 、副次的評価項目である臨床的および電気生理学的評価項目に関して、IVIGと血漿交換の同等の有効性が示された。
今日では、筋無力症クリーゼの治療だけでなく、標準的な免疫抑制療法では病勢コントロールが不十分な患者や禁忌のある患者の維持療法にも、血漿交換の代わりに免疫吸着療法が頻繁に用いられている。免疫吸着療法は、血漿交換療法と比較して、MGの治療に同等の効果があることが示されている。免疫吸着の利点には、血漿蛋白質や凝固因子の代替が不要であること、血漿交換と比較してはるかに多量の血漿 (2-2.5倍) の迅速な治療が可能であることなどがある。さらに、免疫吸着の合併症や副作用は、血漿交換と比較して有意に減少しているようである。
5. 胸腺切除術
MG患者における胸腺切除術は、その他の臨床状況が安定していれば常に行われるべきである。すなわち、GCSや他の免疫抑制療法、かつ/または血漿交換や免疫抑制、IVIGを用いた前処置が効果的に行われている場合には、周術期死亡率は1%未満に低下する。胸腺切除術の臨床的効果は、数年後に現れることが多い。
5-1. 胸腺腫関連MGに対する胸腺切除術
胸腺腫のある症例では、MGの有無やその重症度、またその他の自己免疫疾患の有無にかかわらず、胸腺腫切除の適応がある。
5-2. 非胸腺腫関連AChR抗体関連MGに対する胸腺切除術
胸腺がAChR抗体関連MGの免疫病態発症に関わっていることを考え、胸腺腫は1942年から治療選択肢として選択されてきた。しかし、胸腺腫そのものと標準的免疫抑制治療の効果を比較したランダム化比較試験は現時点では存在しない。現在進行形の非胸腺腫関連AChR抗体関連MGに対する多施設一重盲検ランダム化第3相試験 (MGTX試験; 経胸骨拡大胸腺摘除術と胸腺摘除なしの比較) は、2016年まで行われる予定である。
MGTX試験の結果が利用可能になるまで、胸腺切除は次のような臨床的状況において考慮されるべきである: (a) 胸腺切除術は非胸腺腫関連全身型MGについてはメタ解析のエビデンスが存在するため適応があるかもしれない、(b) 非胸腺腫関連眼筋型筋無力症の患者は臨床試験からの十分なエビデンスが存在しないため単一症例ごとに意思決定される必要がある。胸腺腫のない眼筋型筋無力症の患者に対する胸腺切除術は、全身化を防ぐことができる可能性がある。
15-50歳の胸腺腫を伴わない全身型AChR抗体関連MGに対する発症から1-2年以内の胸腺切除は、有益性が高いように見える。しかし、これらの年齢制限は恣意的であり、一部の専門家はこの制限を厳しく考慮してはいない。胸腺腫のない5-14歳の小児および成人AChR抗体関連MGに関しては、胸腺が未だ免疫システムの発達に関する役割を有していることから、対症療法と免疫抑制治療に十分な反応性がない場合にのみ行われる方がよい。しかし、一部の研究では1.5歳における胸腺切除も免疫機能の障害をきたさないことが示されている。
5-3. 非胸腺腫関連MuSK抗体関連MGに対する胸腺切除術
AchR抗体関連MGとは対照的に、MuSK抗体関連MG患者の胸腺病理は比較的稀である。ある研究では、15人のMuSK抗体陽性患者において胸腺切除術の効果を証明することはできなかったが、別の研究では、MuSK抗体は胸腺切除術の予後不良を予測した。したがって、利用可能なエビデンスからは、一般的にMuSK抗体関連MGでは胸腺切除術を推奨すべきでないことが示唆される。しかし、胸腺切除術後に明らかに改善したMuSK抗体陽性患者はほとんど報告されていないため、疾患のコントロールが不良な個々の症例では、胸腺切除術を考慮してもよいであろう。
5-4. 非胸腺腫性血清反応陰性MGに対する胸腺切除術
胸腺切除術に関する後ろ向きコホート研究では、少なくとも3年間の追跡を行ったAChR抗体陰性およびAChR抗体陽性のMG患者において、同様の術後結果が報告されている。胸腺切除術後の寛解または改善は、AChR抗体陰性患者の57%、AChR抗体陽性患者の51%であった。したがって、胸腺切除術は、AChR抗体陽性患者と同様に、AChR抗体およびMuSK抗体が検出されない全身型MGの患者にも推奨される。
5-5. 胸腺切除術の手技
胸腺切除術の標準的な手技は、最大限の胸腺切除を目指して胸腺全体と後胸骨脂肪組織を切除する経胸骨拡大胸腺切除術であった。しかし、最小侵襲的介入がますます適用されるようになっている。交絡因子が顕著であるため、これらの研究を相互に比較することはできないが、報告されている治療効果や臨床的代替物に対する効果は同等であるようである。したがって、低侵襲胸腺切除術は、ゴールドスタンダードである経胸骨拡大胸腺切除術に代わる選択肢であり、使用する施設が増加している。
推奨される治療レジメンを表3に示す。

6. 結論
MGは自己免疫性の抗体介在性神経筋シナプス伝達障害である。MGの臨床的ホールマークは、変動性のある易疲労性、眼筋、球、四肢 (近医) 骨格筋群の筋力低下である。MGは、明確な免疫遺伝学的特性を伴う自己免疫性疾患として発症するか、胸腺腫瘍に伴う傍腫瘍性症候群として発症するか、のいずれかであると考えられる。どちらのケースでも、中枢性胸腺自己寛容と末梢性自己寛容メカニズムが自己免疫性CD4+ T細胞介在性B細胞活性化を好み、病原性の高親和性IgG1、3、または4サブクラスの合成に進むと考えられる。これらの自己抗体はAChRそのものや、シナプス後膜におけるAChRクラスタリングと神経筋シナプスの構造的維持に関与するMuSK、LRP4、agrinに結合する。
MGの治療戦略は、以下の3つに分類される: (a) 神経筋伝達を促進するAChEIによる対症療法、(b) 急性期治療介入のための抗体除去治療 (IVIG、血漿交換、または免疫吸着)、(c) 維持治療のための免疫抑制治療 (基礎治療としてGCS+AZA、CSA、MTX、MMF、TCM、エスカレーション療法としてCPPやRTX)。その他のモノクローナル抗体、プロテオソーム阻害剤、免疫/骨髄除去療法と造血幹細胞移植は、ごく一部の症例でのみ用いられる。EOMGとTAMGの免疫病態機序における胸腺の役割を考えると、胸腺切除術は十分な臨床的安定が得られたのちに行われるべきである。
感想
病態面白かった。AIREなんて文字列を見たのは学生時代以来だった。
特に、LOMGの免疫学的特徴がTAMGとかなり類似しているというのは興味深かった。発症時の末梢血T細胞レパトアがLOMGとTAMGで類似しているという引用文献の中身を読んでみたけど、これはウイルス感染などを契機にしたクローン性CD8陽性T細胞の増加がepitope spreadingを引き起こして、自己反応性Th細胞を活性化させて抗体産生を招くというロジックだった。EOMGでは胸腺内で抗原提示や抗体産生までの初期段階が完結しているのに対して、LOMGやTAMGでは (負の選択に異常がある?) 胸腺が自己反応性Th細胞を末梢に送り出してしまっている状況下で、感染などをトリガーにして発症をきたしているらしい。じゃあ結局なぜ自己反応性Th細胞が末梢に存在するのかというと、TAMGではそれが胸腺腫のせいとされているけれど、LOMGではよくわかっていなさそう。本文中には発症前に胸腺腫があって発症時には消えているのではないか、とかLOMGでも残存胸腺からT細胞が送り出されているのではないか、などと書かれていたけれど、ほんとかなあ~という雰囲気も否めない。ほんとかもしれないけども。